Our Research
mRNA life cycles are dynamic and diverse, with thousands of mRNAs produced per minute in a typical mammalian cell. Before translation, mRNAs move through subcellular compartments, including chromatin release and nuclear export. In the cytoplasm, ribosomes load onto mRNAs, which are eventually degraded. Numerous regulatory mechanisms control these transitions between compartments.
In eukaryotic cells, nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) encoded RNAs have distinct life cycles. Human mtDNA, originating from a eubacterial genome, has significantly reduced in size due to gene loss or transfer. The remaining genome encodes 13 proteins, all core subunits of the oxidative phosphorylation (OXPHOS) complexes. The other OXPHOS subunits are nuclear-encoded, and the coordinated expression and regulation of nDNA and mtDNA protect against mitonuclear imbalance, where OXPHOS subunits accumulate discordantly. Achieving this balance requires complex regulatory mechanisms that coordinate gene expression across distinct systems, which employ separate machinery and regulatory processes.
We aim to understand how nuclear- and mitochondrial-encoded genes are regulated throughout the life cycles of their transcripts to maintain a balanced proteome. We develop and apply quantitative methods to investigate 1) the control of nuclear-encoded mRNA life cycles and 2) the co-regulation of mitochondrial-encoded and nuclear-encoded OXPHOS genes.

The nuclear mRNA life cycle


Subcellular Timelapse-seq
Our goal is to understand the entire cycle of gene expression. 1) How are pre-mRNAs produced and processed? 2) How do mRNAs avoid nuclear degradation and reach the cytoplasm? 3) How is subcellular localization controlled? We create quantitative methods to examine the complex steps in the mRNA life cycle. We are now tracking the production, processing, and cellular flow of mRNAs with the aim of understanding the determinants of the mRNA life cycle.
1) How are pre-mRNAs transcribed and processed on chromatin?
In human cells, genes contain numerous introns that need to be excised to form a functional mRNA. We developed direct RNA nanopore sequencing methods, such as nanopore analysis of cotranscriptional processing (nano-COP), to study pre-mRNAs processing without the length-associated biases found in techniques using cDNA synthesis and PCR application. We asked when, where, and how introns are excised from the pre-mRNA. We found that 60% of introns are removed co-transcriptionally, while the pre-mRNA is still attached to Pol II. However, splicing catalysis largely occurs after Pol II has transcribed thousands of more nucleotides.
We examined the sequence of intron removal across pre-mRNAs during post-transcriptional processing and found that introns tend to be excised in one or occasionally two preferred orders that were constant across cell types and alternative splicing. In two examples, we found roles for specific cis-elements in controlling splicing order.
Future directions:
1) How fast are RNA binding proteins and spliceosomal subunits loaded onto nascent RNA?
2) How do cis-elements control splicing order?
Read more:
Drexler, H. L., Choquet, K., Churchman, L. S. Splicing kinetics and coordination revealed by direct nascent RNA sequencing through nanopores. Mol. Cell 77, 985–998.e8 (2020).
Choquet, K., Koenigs, A., Dülk, S. L., Smalec, B. M., Rouskin, S., Churchman, L.S. Pre-mRNA splicing order is predetermined and maintains splicing fidelity across multi-intronic transcripts. Nat. Struct. Biol. 30, 1064–1076 (2023).
2) How and why are mRNAs targeted for nuclear degradation?
Textbook views of the mRNA life cycle typically indicate that all mRNAs are exported. However there is a nuclear RNA exosome that degrades introns, antisense RNAs and other non-coding RNAs. We recently found that nuclear mRNA degradation may play a broader role in gene expression regulation than previously appreciated.
Using our recent method, subcellular TimeLapse-seq, we tracked the age of RNAs as they moved across subcellular compartments and determined RNA half-lives at subcellular resolution using a Bayesian modeling framework. Our approach measured the rates at which transcripts were released from chromatin, exported from the nucleus, and loaded onto polysomes for all human mRNAs. We also measured nuclear and cytoplasmic degradation rates.
A simple RNA life cycle model did not fit our data for ~10% of genes. Instead, a model including substantial nuclear mRNA degradation was required. As a result, hundreds of genes were "predicted to undergo nuclear degradation" (PUNDs), where most (>85%) of the transcripts encoded by these genes are never exported from the nucleus. Many PUND genes participate in gene expression and RNA biology, including ribosome subunits, RNA splicing, and nuclear mRNA export factors. PUND transcripts possess unique features like incomplete splicing and long poly(A) tails, which may aid their identification by nuclear degradation pathways. Overall, our data predict pervasive nuclear transcript degradation, potentially playing a role in nuclear RNA homeostasis.
Future directions
1) How are PUND transcripts targeted for nuclear decay?
2) What is the function of nuclear mRNA degradation?
Read more:
Ietswaart R, Smalec BM, Xu A, Choquet K, McShane E, Jowhar ZM, Guegler CK, Baxter-Koenigs AR, West ER, Fu BXH, Gilbert L, Floor SN, Churchman LS. Genome-wide quantification of RNA flow across subcellular compartments reveals determinants of the mammalian transcript life cycle. Mol Cell 2024
3) How is mRNA subcellular localization controlled?
Using subcellular Timelapse-seq to measure the rates of transitions between cellular compartments, we found that RNAs do not flow across the cell at the same rates. Our results demonstrated that functionally related genes undergo similar rates of RNA flow and that the targets of many RNA binding proteins (RBPs) exhibit different RNA flow rates compared to other genes.
Our study revealed wide variations (>100-fold) in RNA flow rates between genes and subcellular compartments. Functionally-related genes experienced similar RNA flow rates, and targets of many RNA binding proteins exhibited different flow rates compared to other genes. We measured poly(A) tails with subcellular resolution and showed that tail lengths reflect subcellular RNA half-lives. We identified genetic and molecular features that predict RNA flow using machine learning, including transcription factors and sequence elements. Our findings uncovered many determinants of mRNA dynamics throughout its life cycle.
Future directions:
1) How do mRNAs flow to organelles and other compartments?
2) How do transcript isoforms impact RNA flow?
Read more:
SIetswaart R, Smalec BM, Xu A, Choquet K, McShane E, Jowhar ZM, Guegler CK, Baxter-Koenigs AR, West ER, Fu BXH, Gilbert L, Floor SN, Churchman LS. Genome-wide quantification of RNA flow across subcellular compartments reveals determinants of the mammalian transcript life cycle. Mol Cell 2024
How do eukaryotic cells coordinate gene expression between two disparate genomes in separate cellular compartments? Our aim is to understand the processes that govern the balance between nuclear and mitochondrial gene expression. We have adapted and applied methods to analyze mtDNA expression quantitatively. Our first studies in this area asked whether nuclear and mitochondrial gene expression processes were coordinated. By adapting ribosome profiling to assess mitochondrial protein synthesis, we demonstrated that the translation of nuclear- and mitochondrial-encoded OXPHOS subunits is balanced in both yeast and human cells.
Expanding on these findings, we are currently investigating the following questions. 1) What are the critical regulatory control points of the mitochondrial genome? 2) How do nuclear-encoded factors and metabolites influence mitochondrial gene expression? 3) How does mitonuclear co-regulation occur in highly polarized cells, like neurons?
1) What are the critical regulatory points of the human mitochondrial genome?
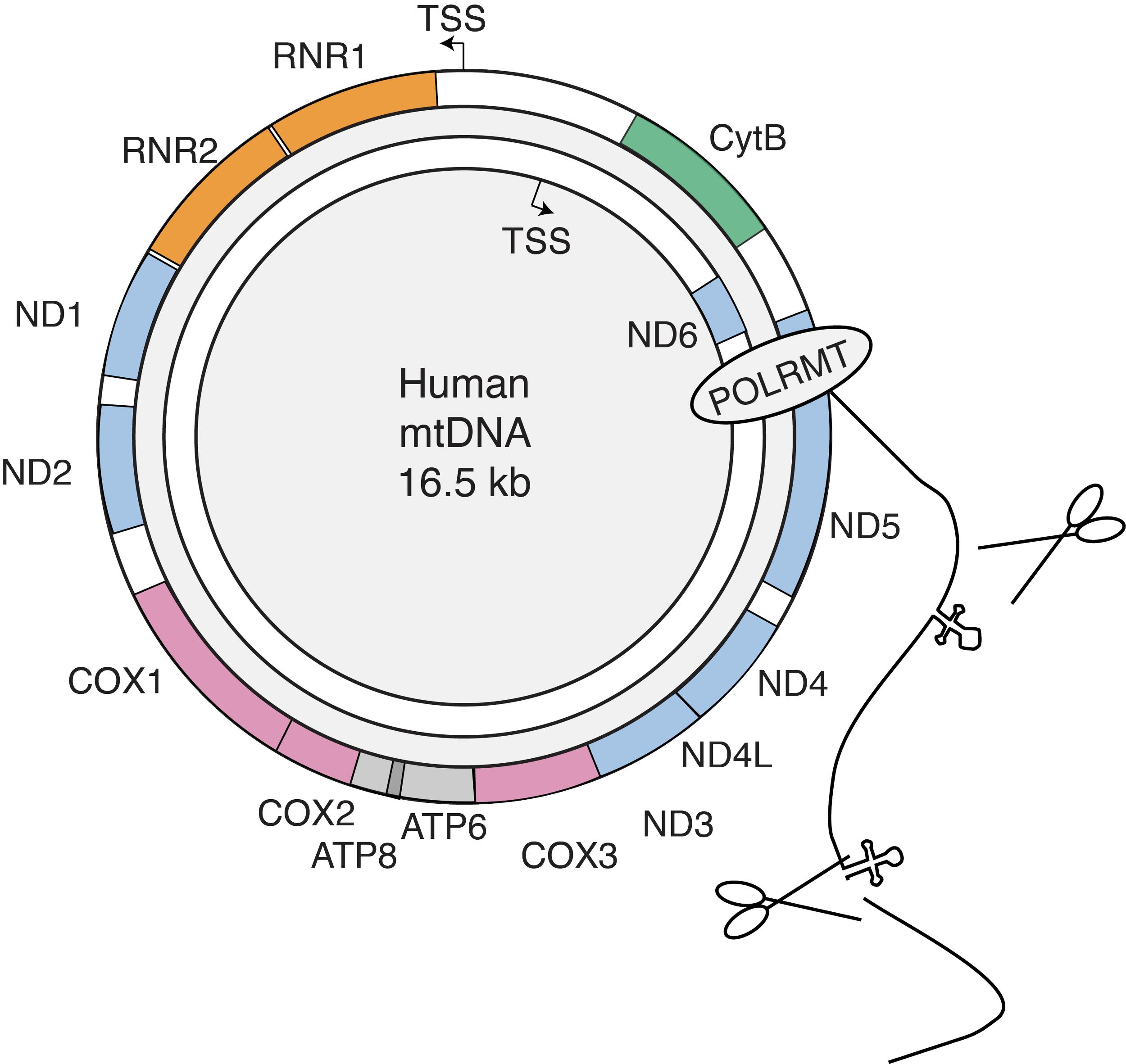
Mitochondrial gene expression in human cells has important characteristics distinct to those of yeast mitochondria and nuclear gene expression. To pinpoint the stages of mitochondrial gene expression that are most regulated, we are quantitatively dissecting each step.
mtDNA packaging. Human cells contain hundreds of mtDNA molecules distributed throughout the mitochondrial network. These molecules, over 5 µm in length, are compressed into 100-nanometer nucleoprotein complexes called nucleoids. We used our single-molecule technique, mtFiber-seq, to measure the accessibility of full-length mtDNA molecules in various human cell types. We found that most nucleoids are inaccessible, and the proportion varied depending on cell type and mitochondrial function. Unlike nuclear genome compaction, mtDNA accessibility is largely all-or-none, suggesting that genome activation may represent an additional layer of gene expression regulation.
The mitochondrial mRNA life cycle. The non-coding region of the 16.5 kb genome contains two promoters that generate two near-genome length polycistronic transcripts, which are then cleaved into individual mRNAs with little to no 5' UTRs. We measured the mt-RNA life cycle (RNA production, processing, ribosome arrival, and turnover) using Timelapse-seq and direct nanopore RNA sequencing to identify regulatory control points in mitochondrial gene expression. By analyzing these data and modeling protein synthesis levels, we discovered that mitochondrial mRNA turnover rates and translation efficiency mainly account for protein synthesis levels in human mitochondria.
Future directions
1) What is the spatial organization of mtDNA regulation and expression?
Read more:
Couvillion, M. T., Soto, I. C., Shipkovenska, G., Churchman, L. S. Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503 (2016).
Soto, I.*, Couvillion, M.*, Hansen, K. G., McShane, E., Moran, J. C., Barrientos, A., Churchman, L. S. Balanced mitochondrial and cytosolic translatomes underlie the biogenesis of human respiratory complexes. Genome Biol. 23, 170 (2022). * denotes co-first authors
Isaac, R. S., Tullius, T. W., Hansen, K. G., Dubocanin, D., Couvillion, M., Stergachis, A. B.#, Churchman, L.S.# Single-nucleoid architecture reveals heterogeneous packaging of mitochondrial DNA. Nat. Struct. Mol. Biol. 31, 568–577 (2024) # denotes co-corresponding authors
McShane, E., Couvillion, M., Ietswaart, R., Prakash, G., Smalec, B. M., Soto, I., Baxter-Koenigs, A. R., Choquet, K., Churchman, L. S. A kinetic dichotomy between mitochondrial and nuclear gene expression drives OXPHOS biogenesis. Mol Cell, 2024
2) How do nuclear-encoded factors and metabolites influence mitochondrial gene expression?
Discovered by genetic screens, yeast translation activators (TAs) are nuclear-encoded and imported into mitochondria to promote the translation of specific mRNAs via their 5' UTRs. To identify human genes that control mitochondrial gene expression, we developed genome-wide CRISPR screen strategies to identify factors that affect the balance of Complex IV subunits, mitochondrial-encoded COX1 and nuclear-encoded COX4. We identified known regulators of mitochondrial gene expression, such as TACO1, whose mutations cause Leigh syndrome, a metabolic disorder, and is essential for COX1 synthesis. Among the genes with unclear mitochondrial functions, we are initially focusing on PREPL, a protein highly expressed in the brain, and NME6, a member of the nucleotide diphosphate kinase (NDPK) family.
Future directions
1) How is mitochondrial translation regulated?
2) Do mitochondrial metabolite pools influence mitochondrial gene regulation?
Read more:
Kramer, N. J.*, Prakash, G.*, Choquet, K., Soto, I., Petrova, B., Merens, H. E., Kanarek, N., Churchman, L.S. Regulators of mitonuclear balance link mitochondrial metabolism to mtDNA expression. Nat. Cell Biol. 25, 1575–1589 (2023). * denotes co-first authors
3) How does mitonuclear co-regulation occur in neurons?
Neurons are highly dependent on mitochondrial function, and their complex, polarized morphology offers a challenge for balanced nuclear and mitochondrial gene expression. We aim to determine if this structure makes neurons more susceptible to mitonuclear imbalances due to genetic or physiological stress.
Future directions
1) Is nuclear- and mitochondrial-encoded gene expression balanced in neurons?
2) How does mitonuclear balance vary between soma and neurite-localized mitochondria?
3) Is this relevant to mitochondrial malfunction in neurodegenerative diseases?
Mitonuclear co-regulation